
Genome based LAMP primer design for a set of target genomes
I. Introduction
LAMP(Loop-mediated isothermal amplification) is a simple and effective method to amplify DNA sequence.A primer set is required for LAMP. One LAMP primer set contains four single LAMP primers from six primer regions in a genomic region. The six regions are called F3, F2, F1c, B1c, B2 and B3 regions. Sequences from F1c and F2 regions are synthesized into a primer called FIP and sequences from B1c and B2 regions are synthesized into a primer called BIP. In order to accelerate the amplification, two additional loop primers(LF, LB) can be added.
The positional relationship among those regions are shown as follows:
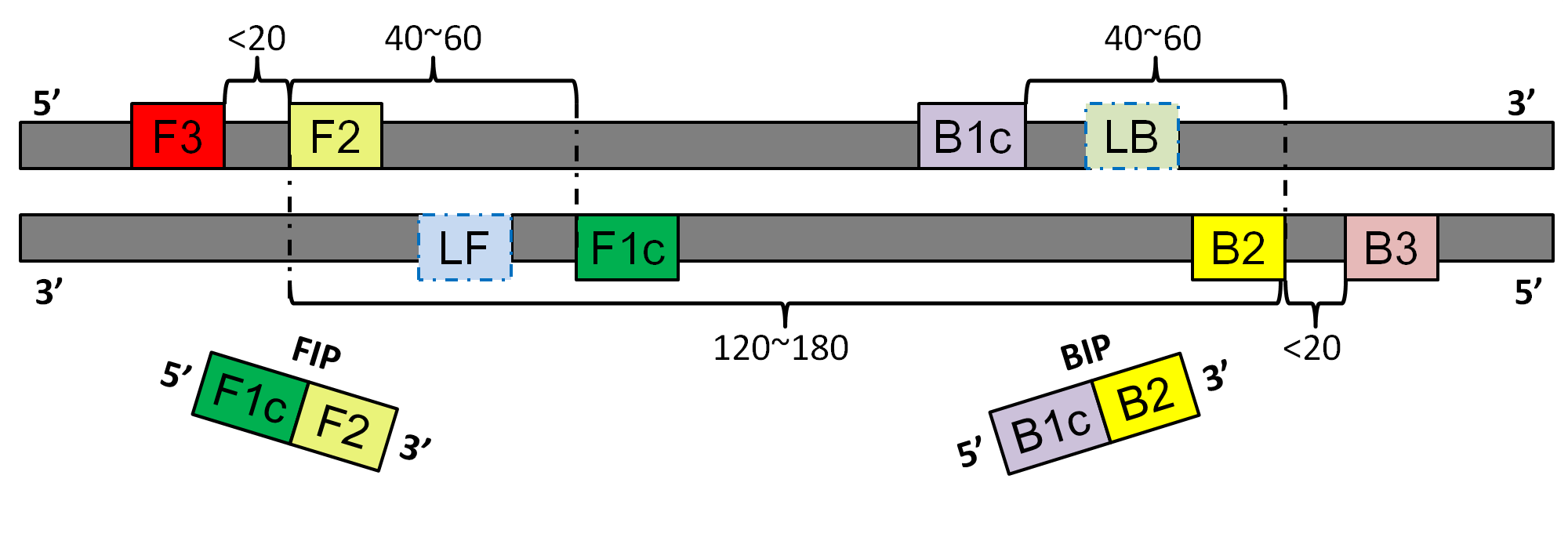
GLAPD can design LAMP primer sets based on a whole genome. It can also design LAMP primers for a set of target genomes. Users can specify a group of background genomes.
Firstly, GLAPD identifies all candiate single primer regions. Then those single primers are combinded into LAMP primer sets. Thirdly, the LAMP primer sets were aligned to the target genome and all background genomes and only those primer sets for the target genomes and only these genomes are output.
II. System requirements
GLAPD runs under Linux operation system. It needs perl and gcc. For the GPU version, the computer needs GPUs installed >=5.0.
Other software needed are listed below:
Bowtie, which could be downloaded from http://bowtie-bio.sourceforge.net/index.shtml. GLAPD contains the binary bowtie program.
CUDA driver, which could be downloaded from http://www.nvidia.com.
III. Installation
1. Download:
A tar ball for GLAPD system (v0.2, 02/12/2026) can be downloaded here
2. Decompressing the files: tar -zxvf GLAPD.tar.gz
3. Compile: 3.1. If you want to use the CPU version:
cd GLAPD/
make
3.2. If you want to use the GPU version:cd GLAPD/GPU/
make
IV.Quick start
An example is provided below to show how to run GLAPD.
1. If you want design LAMP primers for a sequence without taking care of commonality and specificity:
cd GLAPD/
./Single -in example/example.fa -out Test
(Two files "Inner/Test" and "Outer/Test" are created.)
./LAMP -in Test -ref example/example.fa -out success.txt
(Ten LAMP primer sets are designed successfully stored in "success.txt" file.)
2. If you want design common LAMP primers without taking care of specificity:
cd GLAPD/
./Single -in example/example.fa -out Test
(Two files "Inner/Test" and "Outer/Test" are created.)
perl par.pl --in Test --ref example/example.fa --bowtie Bowtie_path/bowtie --index example/index --common example/target-list.txt
(Three files "Inner/Test-common_list.txt", "Inner/Test-common.txt" and "Outer/Test-common.txt" are created.)
./LAMP -in Test -ref example/example.fa -out success.txt -common
(Ten LAMP primer sets are designed successfully stored in "success.txt" file.)
3. If you want design specific LAMP primers without taking care of commonality:
cd GLAPD/
./Single -in example/example.fa -out Test
(Two files "Inner/Test" and "Outer/Test" are created.)
perl par.pl --in Test --ref example/example.fa --bowtie Bowtie_path/bowtie --index example/index --specific example/background-list.txt
(Two files "Inner/Test-specific.txt" and "Outer/Test-specific.txt" are created.)
./LAMP -in Test -ref example/example.fa -out success.txt -specific
(Ten LAMP primer sets are designed successfully stored in "success.txt" file.)
4. If you want design common and specific LAMP primers:
cd GLAPD/
./Single -in example/example.fa -out Test
(Two files "Inner/Test" and "Outer/Test" are created.)
perl par.pl --in Test --ref example/example.fa --bowtie Bowtie_path/bowtie --index example/index --common example/target-list.txt --left
(Five files "Inner/Test-common_list.txt", "Inner/Test-common.txt", "Inner/Test-specific.txt", "Outer/Test-common.txt" and "Outer/Test-specific.txt" are created.)
./LAMP -in Test -ref example/example.fa -out success.txt -common -specific
(The first ten LAMP primer sets met all requirements are stored in a file called "success.txt".)
V.Usage of GLAPD system
There are three steps to run GLAPD. The usage of each step was listed below.
1.Identifying candidate single primer regions:
Command:
Single -in <ref_genome> -out <single_primers> [options]*
Arguments:
-in <ref_genome>
the reference genome in FASTA format.
-out <single_primers>
output the candidate single primers
-dir <directory>
the directory for output file
default is the current directory
-loop
identifying candidate single primer regions for loop primers
-check <int>
check the secondary structure of the single primers or not
0: don't check the secondary structure; other values: check the secondary structure
by default: this value is set to 1. the secondary structure will be checked for all primers.
-par <par_directory>
parameter files under the directory are used to check primers' secondary structure
default: GLAPD/Par/
-h[-help]
print usage
2.Aligning sequences from single primer regions(optional):
Command:
perl par.pl --in <sinlge_primers_file> --ref <ref_genome> --common[--specific] <genomes_list> --bowtie <bowtie> --index <database> [options]*
Arguments:
--in <single_primers_file>
the file name of candidate single primer regions, files are generated from Single program
--ref <ref_genome>
reference genome, fasta formate
--dir <directory>
dirctory for files of candidate single primer regions
default: current directory
--loop
include loop primers
--common <genomes_list>
the genomes in the file(target genomes) are expected to be amplified by LAMP primer sets
--specific <genomes_list>
the genomes in the file(background genomes) are not expected to be amplified by LAMP primer sets
--left
background_group = all_genome_in_database - target_group
used with --common
invalid if exist --specific
--bowtie <bowtie>
the bowtie program
--index <database>
bowtie index file name, comma-separated
--mis_s <int>
the max number of mismatches allowed when align single primers to background genomes
this value between 0 and 3. the bigger of the value, the more specific
default: 2
--mis_c <int>
the max number of mismatches allowed when align single primers to target genomes
this value between 0 and 3. the smaller of the value, the more common
default: 0
--threads <int>
number of threads to launch when align
default: 1
--help|--h
print help information
3.Designing LAMP primer sets:
Command:
LAMP -in <sinlge_primers_file> -ref <ref_genome> -out <LAMP_primer_sets> [options]*
Arguments:
-in <single_primers_file>
the file name of candidate single primer regions, files are generated from Single program
-ref <ref_genome>
reference genome, fasta formate
-dir <directory>
the directory for output file
default: current directory
-out <LAMP_primer_sets>
output successfully designed LAMP primer sets
-num <int>
the expected output number of LAMP primer sets
default: 10
-loop
design LAMP primer sets with loop primers
-common
design common LAMP primer sets those can amplify more than one target genomes
-specific
design specific LAMP primer sets those can't amplify any background genomes
-check <int>
check primers' tendency of binding to another in one LAMP primer set or not
0: don't check; other values: check
default: 1
-par <par_directory>
parameter files under the directory are used to check primers' binding tendency
default: GLAPD/Par/
-fast
fast mode to design LAMP primer sets. With this mode, GLAPD may lost some results
-h/-help
print usage
VI.Tips:
1) Selecting the reference genome:
The reference genome can be selected randomly from the group of target genomes, or the most expected genome targeted by the LAMP primer set can be selected.
2) Configuration files:In step 2, if you have a file (called "common file") containing the list of genomes with common regions, you can use the "--left" option to replace the "--specific" option. In this way, all genomes in database exept those listed in this "common file" will be defined as the background genomes.
3) Fast mode:In step 3, there is a "-fast" option to accelerate the designing procedure. The program will stop when enough number of primer sets have been designed. Therefore, this mode may miss some best LAMP primer sets.
Citation:
If you use GLAPD, please cite our paper below. Contact:
If you have any question, please feel free to contact us.
Ben Jia: chenmodexiaoxi@126.com
Chaochun Wei: ccwei@sjtu.edu.cn
Please send your comments or bug reports to Dr. Wei .