De novo assembly strategies
EUPAN provides an optimized strategy (linearK) for de novo assembly with iterative use of SOAPdenovo2. The key idea of this variant method is to select the best Kmer for each sample. This strategy shows both relative good speed and assembly performance. Here we describe the strategy in detail.
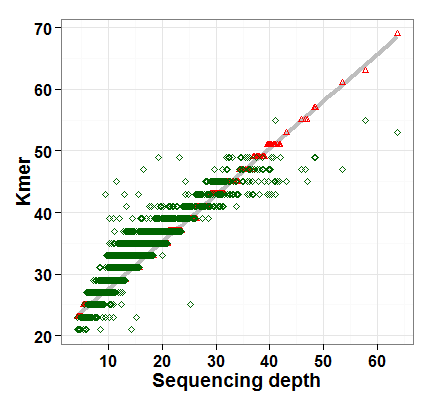
The basis of this strategy is that the best Kmer (evaluated by the N50 of assembly results) and the sequencing depth are linear correlated (see example Figure A). Therefore, we can estimate the linear function with a small number of individuals (e.g. 30) and then applied the function to hundreds even thousands of individuals.
In EUPAN, the assembly algorithm is as follows (Figure B). We first determined an initialized Kmer for rice accession R based on a linear model $$ K_{init} = 2 * \lfloor 0.5 * (a * Dept + b) \rfloor + 1 $$ where Dep is the sequencing depth of rice R and a and b are the parameters of the linear model. After Kinit is determined, we run SOAPdenovo 3 times with 3 different Kmer with a step of 2 ($ K_{low} = K_{init} - 2$, $K_{mid} = K_{init}$ and $K_{high} = K_{init} + 2$). N50 of all assemblies were calculated and were compared with each other. If N50 of $K_{mid}$ is the highest, assembly at $K_{mid}$ is selected as the final result and program stops. If N50 of $K_{high}$ is the highest, an additional run with $Kmer=K_{high} + 2$ is needed and we compared N50 of the 3 assemblies; this iteration continuous until N50 of the median Kmer is the highest. If N50 of Kmerlow is the highest, Kmer goes down and similar iteration is carried out to determine the best Kmer.

Therefore, to use this optimized strategy in EUPAN, the user should first selected a subset of the individuals with diverse sequencing depths for training the linear function. For training purpose, please set an unlimited number of iteration times (-u option of “assemble linearK” tool, -u 100). After training, the user should find the best Kmer for each sample in the output directory and a linear fit is needed to calculated the parameter a and b of the linear function. Now we are ready for large-scale assemblies. At this time, please use the –r option of “assemble linearK” tool to provide the parameter of the linear functions to the program and set the -u option to a small value (e.g. –u 10). Moreover, please set the genome size (-g option) and sequencing properties (-c option) properly for both the training assemblies and large-scale assemblies.
We have tested this strategy in 3000 Rice Genomes Project. The linear model were trained on 50 randomly selected rice accessions and then applied to assemblies of over 3,000 rice accessions. On average, ~3.94 times of running SOAP denovo were used for each sample.