The human reference genome is still incomplete, especially for those population-specific or individual-specific regions, which may have important functions. It encourages us to bulit the pan-genome of a human population. Previously, we developed a "map-to-pan" strategy - EUPAN, specific for eukaryotic pan-genome analysis. However, due to the large genome size of individual human genome, EUPAN is not suit for pan-genome analysis involving in hundreds of individual genomes. Here, we present a improved tool, HUPAN (HUman Pan-genome ANalysis), for human pan-genome analysis.
We propose HUPAN strategy primarily in the 185 deep sequencing and 90 assembled Han Chinese genomes. HUPAN uiltized all the well-conceived strategies of EUPAN. Besides, HUPAN has a number of distinct improvements as follows:
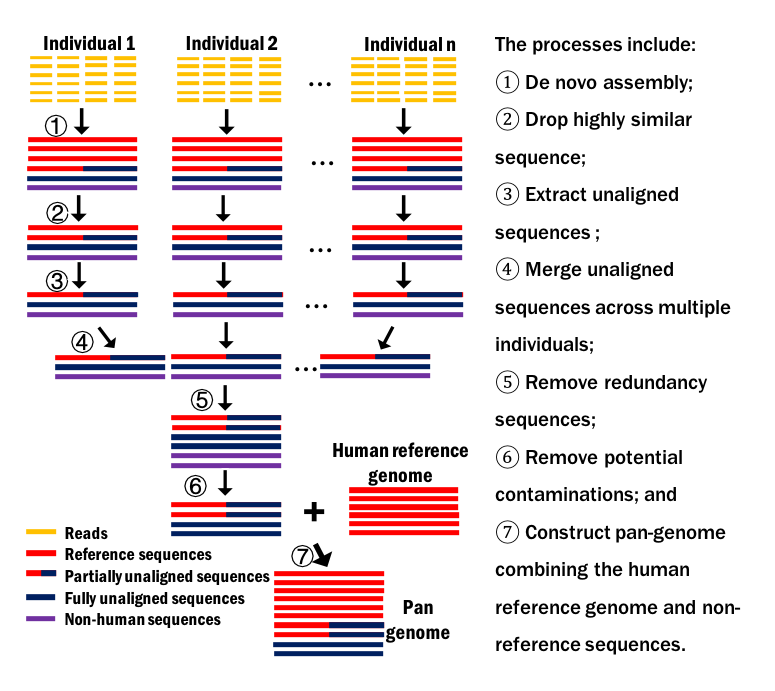
- De novo assembly of individual genome is preformed with low memory;
- Fast extracting non-reference sequences of larger genome is achieved;
- Considering both fully unaligned sequences and partilly unaligned sequences;
- A rigorous screening process is proposed to distinguish non-human sequences from non-reference sequences;
- Novel protein coding genes on non-reference sequences were predicted and analyzed.
Human genome studies always involve big data and various softwares; and require very careful parameter selection process. Therfore HUPAN toolbox provides 3 types of tools: 1) single machine version, 2) LSF version (working on supercomputer based on LSF system, in which, "bsub" is used to submit jobs) and 3) SLURM version (working on supercomputer based on SLURM system, in which, "sbatch" is used to submit jobs).
Use & Citation
HUPAN is free for non-commercial use (CC BY-NC 4.0). For commercial use, please contact the authors.
Contact Information
Zhongqu Duan: zhqduan@sjtu.edu.cn
Chaochun Wei: ccwei@sjtu.edu.cn
News
- 2019.3.21 version 1.02 released.
- Fix several known bugs.
- Add a novel tool ---- "filterNovGene", to filter the novel precited genes.
- 2018.11.28 version 1.01 released.
- The "getUnalnCtg" and/or "mergeUnalnCtg" tools are optimized to collect and merge both fully unaligned contigs and partilly unaligned contigs from individual quast result. In addition, the coordinates of partially unaligned contigs on reference genome are also collected.
- Add a novel tool ---- "blastAlign", to align sequences to target sequences by blast.
- Add a novel tool ---- "getTaxClass", to obtain the taxonomic classification of contigs based on the accession id in blast result.
- Add a novel tool ---- "rmCtm",to detect and discard the potentail contamination.
- Add a novel tool ---- "splitSeq", to split sequence file into multiple small size files.
- Add a novel tool ---- "genePro", to ab initio gene predict on the non-reference sequences.
- Add a novel tool ---- "mergeNovGene", to merge results from multiple maker result files.
- Add a novel tool ---- "simSeq", to simulate and plot the total size of novel sequences.
- 2018.9.10 version 1.00 released.
- "assembly" tool now can perform de novo assembly with low memory.
- Add a novel tool ---- "alignContig", to align the assembly results to reference genome.
- Add a novel tool ---- "extractSeq", to extract lower similarity sequences.